USP <382>: Think Systems, Not Components - Fragmentation, Plunger and Tip-Cap/Needle-Shield Functional Suitability of Syringes and Cartridges – Part 3
United States Pharmacopeia (USP) requirements for elastomeric components of container closure systems used for parenteral products are changing. The original USP chapter for elastomeric components used in injectable drug packaging was <381> Elastomeric Components in Injectable Pharmaceutical Production Packaging/Delivery Systems, which addressed characterizing for biological reactivity, physicochemical testing and functionality testing. The new chapter, USP <382> Elastomeric Component Functional Suitability in Parenteral Product Packaging/Delivery Systems, will become effective on December 1, 2025 and will update the functionality tests for elastomers, whereas the functionality testing portion of USP <381> will be omitted. While under USP <381> compliance was typically a supplier responsibility, under the new chapter it will be the drug manufacturer’s responsibility to meet the functionality requirements on the final system, including the container closure system and the drug.
![]()
USP <382> focuses on vial and bottle packaging, blow-fill-seal (BFS) packaging, plastic packaging (including film bags and blow-molded containers), cartridge packaging systems and syringes (both pre-filled and single-use) and applies to all elastomeric components in direct and indirect contact with the drug product in primary packaging and combination product kits. The USP chapter specifically states that, “[t]he tests for functional suitability described in this chapter are intended to evaluate the fitness of an elastomeric component as part of a specific, final, parenteral product packaging/delivery system.” This is a change from USP <381> in that the whole container-closure system must be certified for functionality rather than simply the individual component. The USP chapter describes this type of holistic testing as “fitness-for-intended-use functional suitability.” As such, the elastomeric component manufacturer cannot perform USP <382> testing without knowledge of the specific drug product/placebo or the complete packaging system that the drug manufacturer plans to use.
The USP chapter prescribes specific testing to be performed for the packaging/delivery system as a starting point but warns that the prescription is not exhaustive and should be used as appropriate, with justification. The chapter further states that functional suitability assessment studies for each specific drug product in its final drug packaging/delivery system should be developed based on a) the system design/mechanics, b) the nature of the dosage form, c) the environment and clinical use setting of the finished drug product and d) the assessed safety of those using and/or exposed to the drug during administration, all with justification. In other words, the USP instructs the drug manufacturer to perform a risk assessment to determine the best testing strategy for the given application.
The present blog focuses on the application of USP <382> prescribed testing to cartridges and syringes, from a mechanical testing perspective. Part 4 of the blog by Jen Roark, USP <382>: Think Systems; Not Components - Packaging/Delivery System Integrity and Needle Self-Sealing Capacity of Syringe and Cartridge Systems, discusses how USP <382> addresses packaging integrity testing and needle self-sealing capacity for cartridges and syringes. Related blogs, Part-1 and Part-2 in this series, focused on the mechanical functionality and system integrity of vial systems.
Cartridges (Figure 1) are used in devices like pen injectors, dental local anesthesia products and on-body delivery devices. They are comprised of a glass- or polymer-based body, an elastomeric plunger, an elastomeric septum, and a seal, which is typically an aluminum ferrule. The septum is intended to be punctured by a double-sided needle. The plunger is intended to be pushed by a motor-driven piston (in an on-body device) or a manually-driven piston (in a pen-injector or dental anesthesia product). West has extensive articles on these components in the Knowledge Center, such as this study.

Figure 1: Prefilled Cartridges
Prefilled syringes (Figure 2) are filled by the manufacturer and are sold in a ready-to-administer, or nearly ready, configuration. They are comprised of a glass- or polymer-based body, an elastomeric plunger (either screwed onto a plunger rod or ready to be screwed onto a plunger rod), either an attached needle or a connector end (e.g., Luer or Luer Lock) and an elastomeric needle-shield or tip-cap which seals the front end of the syringe. The plunger is intended to be pushed by a mechanical injector or manually depressed by a user. Similarly, single use syringes are sold empty but are intended to be used to withdraw drug product from another container, by pulling back on the plunger, and either transfer the drug directly to a patient or to an intermediate device, like an infusion bag.
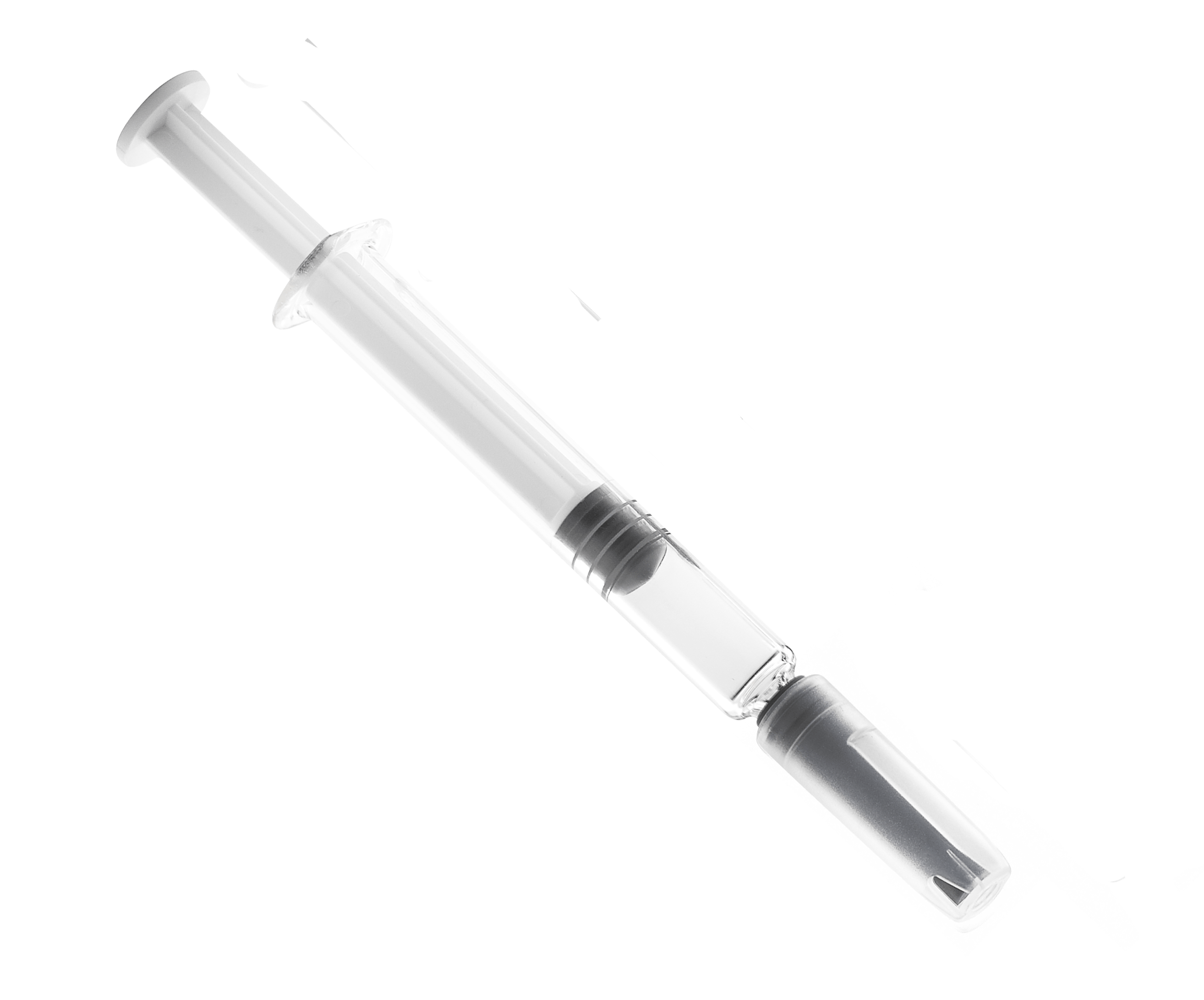
Figure 2: Prefilled syringes
The test samples to be used for the USP <382> testing are intended to closely resemble the final drug product packaging configuration. For cartridges and syringes, that should include identical plungers, bodies, lined seals, tip-caps, needle shields, sample processing (e.g., sterilization) and sample handling (e.g., storage conditions and aging). Depending on the risk assessment of the packaging configuration, one may want to perform ASTM D4169, Standard Practice for Performance Testing of Shipping Containers and Systems, transportation conditioning on the samples prior to testing to assure that samples truly represent what may be used in the field. In contrast, USP <381> simply required that elastomer samples be placed in water, autoclaved at 120oC for a period of time, cooled and air-dried.
Mechanical testing for cartridge and syringe packaged drug products described in the chapter involves testing of fragmentation, plunger functional suitability and tip-cap/needle-shield functional suitability.
Fragmentation
When a cartridge septum is penetrated by a double-sided needle, it sometimes results in fragmentation or coring. This is a topic that was extensively studied at West. Fran DeGrazio described the fundamentals that result in this phenomenon and West’s Knowledge Center has a presentation on factors that contribute to this issue. Since the septum of a cartridge in a multi-use pen-injector is intended to be penetrated multiple times, there is an increased possibility that fragments will be generated by the time that the last dose is dispensed.
To test fragmentation on cartridges, one selects a set of water-filled cartridges and pierces their septa with the needles that are intended for use with that application. For dental local anesthesia cartridges, 12 samples are to be tested, penetrating each septum at least four times, or more if that is the intended use. The needle lumen is to be flushed, after each penetration, onto a filter to collect any fragments caught in the needle and, after completion of the test, the cartridge contents are to be emptied onto the same filter to collect any fragments that ended up in the cartridge. The filter is to be examined under a microscope at 40-to-110-fold magnification, using lighting conditions and equipment as described in USP <788> Particulate Matter in Injections, reporting all particles greater than 150 microns in any dimension. This is a major change from USP <381>, where particle counting was typically performed with the naked eye and limited the count to particles greater than 50 microns in diameter.
For pen-injector cartridges, a similar procedure is specified, except that the total number of penetrations is to equal 100, which can mean 10 cartridges penetrated 10 times each or any other sample/penetration combination, as long as at least 5 samples are used. The key is to match the number of penetrations of each cartridge to the product-use recommendations. If the device would use a new needle for each application, then a new needle must be used for each test penetration.
Break-loose and Extrusion and Plunger Seal Integrity
Plunger functional suitability tests examine the Break-loose force, the Extrusion force and the Plunger Seal Integrity of the cartridge or syringe. When pressure is first applied to a plunger, there is static friction that must be overcome before the plunger will start to move. The force required to initiate this movement is the Break-loose force. Once the plunger starts to move, the force required to overcome the dynamic friction of the plunger moving through the barrel of the cartridge or syringe, plus the force required to push the drug product to flow through the narrow bore of the orifice, is the Extrusion force, or, in the case of empty cartridges and syringes, the Glide force. High friction at this stage results in chattering of the plunger as it exhibits stick-slip behavior. The drug itself would have an impact on this test. Different physical properties of the drug would impact flow through the orifice and therefore testing must be performed with the drug itself or a suitable proxy. West has written about Break-loose and Extrusion/Glide testing in the Knowledge Center. If there is excessive resistance to drug flow through the needle, pressure inside the vessel can induce the drug to flow through alternate paths, such as between the barrel and plunger or past the septum seal, to result in leakage. Testing for this leakage is called the Plunger Seal Integrity test.
To test Break-loose and Extrusion, samples of the container filled with drug product or proxy are to be placed in a mechanical testing machine that can measure applied forces. Those samples which are not equipped with needles must be attached to whatever “connecting devices” the intended use would entail. Forces are to be applied to the plunger at the speed at which they would be applied during intended use and those force measurements are to be recorded. Force specifications are not provided because the anticipated specification would differ for each application and must be determined by the drug manufacturer through risk assessments and human factors studies.
To test the Plunger Seal Integrity of the cartridge or syringe, the container, filled with drug product or proxy, is to be placed in a mechanical testing machine. If the syringe has a fixed needle, or open nozzle, that orifice must be sealed. A force is to be applied to the plunger for a period of time and the sample is observed for evidence of leakage. A dye may be added to the drug product to aid in observation of leakage. The force and duration is either to be based in the intended use of the cartridge or syringe or is to be based on the specification listed in the chapter.
Tip-Cap/Needle-Shield Removal
The distal end of a syringe is either closed off with a tip-cap or with a needle-shield, depending on the type of syringe. Tip-caps can be either Luer (pull-off) or Luer Lock (twist-off). If the tip-cap or needle-shield is too difficult to remove, the syringe cannot be used and the patient would not receive the necessary drug. If the removal is too easy, the cap or shield can fall off, resulting in contamination and/or product loss. The syringe is to be placed in a mechanical testing machine capable of measuring force or torque, as appropriate, and the required force/torque is to be measured at a set axial or rotational speed. Acceptance criteria are to be determined by the drug manufacturer through risk assessments and human factors studies.
West Analytical Laboratory Services can help drug manufacturers navigate the requirements being imposed by USP <382> and perform the necessary testing to assure that products are compliant with the latest regulations. Contact us to learn more about how West can help you with your USP <382> testing needs before the December 1, 2025 deadline.
References:
- USP <381> Elastomeric Components in Injectable Pharmaceutical Product Packaging/Delivery Systems; United States Pharmacopeia
- USP <382> Elastomeric Component Functional Suitability in Parenteral Product Packaging/Delivery Systems; United States Pharmacopeia
- USP <788> Particulate Matter in Injections; United States Pharmacopeia
- ASTM D4169: Standard Practice for Performance Testing of Shipping Containers and Systems


