We apologize your item could not be found. Not all items are listed on our website. Please contact your account manager for details regarding your searched item.
Material number (eg: xxxxxxxxxxx) no space or hyphen needed.
No Results Found
Your Password has changed! Please login again
Cart
Outdated Browser Detected
Our website has detected that you are using an outdated browser that will prevent you from accessing certain features.
Please use one of the below recommended browsers to improve your browsing experience
Evaluation and Identification of Subvisible Particulate Matter in Injections
The presence of particulate matter (PM) in parenteral drug products is a well-known challenge for pharmaceutical companies due to the potential quality and safety risks involved. In biotherapeutics, critical attributes related to efficacy, potency, clinical safety, and immunogenicity can be affected by PM.1-3 PM is defined as extraneous mobile undissolved particles (excluding gas bubbles) that are unintentionally present in solutions.4 Several USP chapters describe the measurement of PM, namely:
<787> Subvisible Particulate Matter in Therapeutic Protein Injections
<788> Particulate Matter in Injections
<789> Particulate Matter in Ophthalmic Solutions
<790> Visible Particulates in Injections
In this article, the scopes of <787> and <788> will be discussed since both chapters address injections and have the same regulatory requirements according to their nominal volumes for subvisible PM.
As compared to visible PM, subvisible PM is unnoticeable without a microscope and is harder to detect. Subvisible particulates present a greater challenge for detection and higher risks that they might go undetected due to their smaller size. As a result, establishing PM profiles for subvisible particulates is highly demanding, and made more so by instrumental limitations and sample handling challenges (to be discussed further in the article).
The occurrence of subvisible PM can stem from several different sources. Particularly, the subvisible particulates found in sterile drug products originating from packaging components and delivery systems are a major concern for drug manufacturers, with potential sources attributed to material components, process aids used to assemble the system, secondary packaging, insufficient washing, and contamination during fill-finish process.5 Therefore, understanding the method of determination and enumeration are critical.
What are Subvisible Particulates and Their Sources?
Subvisible particulates are particles < 100 µm in size and are not visible to the naked eye.6 A related chapter in USP <1787> Measurement of subvisible particulate matter in therapeutic protein injections categorizes this undesired PM as intrinsic, extrinsic, or inherent.
Intrinsic PM arises from sources related to formulation ingredients, packaging, manufacturing processes and equipment. This may also stem from the poor selection or cleaning of components and systems. An example is silicone oil, which is an important manufacturing and product constituent that may ultimately affect PM count.
Environmental factors, that is, materials that are not part of the formulation, package, or assembly process, are the main sources of extrinsic PM. The production environment, including personnel gowning and behaviours can contribute extrinsic PM to the filled product.
Additionally, some products contain a small level of unintended PM that is inherent to the product and therefore, are considered potentially acceptable characteristics of the product. Proteinaceous aggregates are examples of inherent PM found in therapeutic protein drugs, formed by interactions of the protein with itself or with other formulation ingredients. Heterogeneous particulates consisting of more than one chemical entity may also result from protein aggregation. They can be classified as extrinsic or intrinsic based on the nonproteinaceous component that the protein combines with to form the aggregate.
Intrinsic and extrinsic particulates account for the majority of the PM originating from elastomeric closure components and their manufacturing process. Different types of PM have diverse effects on safety and product stability. It is critical to perform PM evaluations and concentration measurements to understand the nature of the particulates involved. Thereafter, the amount of PM can then be minimized in the final drug product upon PM identification and ascertaining their sources.
Determination of PM
USP <787> and <788> describe two procedures for measuring subvisible PM in parenteral products by direct testing of the drug product itself: light obscuration (LO) particle count test and microscopic particle count test.
USP <1788> suggests the use of Flow Imaging (FI) method, not only to complement the above methods, but also allows particles to be characterized into categories of intrinsic, extrinsic and inherent, in the case of biologics for the purpose of risk assessment and continuous improvement. For the simplicity of this article, FI will not be discussed.
LO Particle Count Test vs Microscopic Particle Count Test
An LO particle count test is based on the principle of light scattering that enables automatic determination of particles size and the number of particles according to size. A laser beam that is shone through the liquid sample as it is drawn though an optical flow cell, will be scattered or absorbed by any particles, air bubbles or liquid droplets, thus reducing the total transmitted light. The scattering pattern or “shadow” produced on the light-sensitive detector can then be translated to information on particle size and quantity.
A microscopic particle count test is an alternative technique to LO for subvisible particle analysis. It involves the use of a suitable binocular microscope, a filter assembly for the retention of particulate matter, and a membrane filter for examination. The liquid sample is filtered and inspected through the microscope, followed by manual counting of particles above the size threshold. However, liquid droplets and aggregated proteins that can pass through or absorb onto the filter are not reliably counted by this method.
As a fully automated method, LO can analyze large volumes of samples quickly and easily, with high resolution, minimal operator errors, and without requiring interpretation of data. Therefore, LO is generally the preferred method and the microscopic particle count test is applied only when the former is not applicable. For example, drug products (i.e., emulsions, colloids and liposomal preparations) with high viscosity and/or high opacity are not suitable for LO method. Products that generate air or gas bubbles upon drawing into the sensor are also more appropriate for microscopic particle count testing. This is because these bubbles might be detected as a particle, resulting in false positive data.
As for the microscopic particle count test, complete sample measurement is performed by filtering through the entire sample. The PM retained on the membrane can be used for characterization and identification, if desired (LO method does not allow sample to be reused for further characterization after the count test has been conducted). However, this process requires analyst expertise and experience.
Due to the inherent limitations in particle count techniques, it may be beneficial to utilize both test methods to accurately quantify the number of particles present.
Regulatory Requirements according to USP <787> and <788>
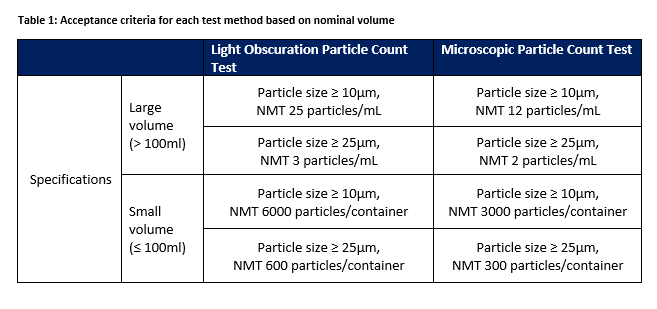
With the different test methods there are different sets of specifications for parenteral infusion or solutions for injection supplied in containers according to volume. The acceptance criteria are illustrated in Table 1 based on each method and the nominal volume of contents within the containers supplied.
West as a Scientific Destination
Understanding of the relevant particle count technology, method capability, and the particle source is critical in mitigating the risks associated with subvisible PM. To achieve accurate and reliable data on subvisible particle count, West can provide comprehensive guidance on the use of analytical methods described in USP <787> and <788>, as well as additional particle characterization methods described in USP <1787> by orthogonal techniques. West’s Analytical Laboratory is capable of performing characterization and identification of particles and this can be performed as part of the development phase, root cause analysis for nonconformance investigations, stability study, and risk assessment.
Information and data obtained from these testing methodologies will aid in the selection of proper components suitable for each application. West is equipped with the appropriate experience, knowledge, and facilities to perform evaluations according to the standards cited and offers testing through its Integrated Solution platform. `
References
Walpot H, Frank RP, Burchard WG, Agternkamp C, Muller FG, Mittermayer CKG. Particulate contamination of intravenous solutions and drug additives during long- term intensive care. Anaesthesist. 1989;38:617-621.
Langille SE. Particulate matter in injectable drug products. PDA J Pharm Sci Technol. 2013;67(3):186-200.
Bukofzer S, Ayres J, Chavez A, et al. Industry perspective on the medical risk of visible particles in injectable drug products. PDA J Pharm Sci Technol. 2015;69(1):123-139.
USP <788> Particulate Matter in Injections. Rockville, MD: US Pharmacopeia.
Rech J, Fradkin A, Krueger A, et al. Evaluation of particle techniques for the characterization of subvisible particles from elastomeric closure components. J Pharm Sci. 2020;109:1725-1735.
USP <1788> Methods for Detection of Particulate Matter in Injections and Ophthalmic Solutions. Rockville, MD: US Pharmacopeia.